Variant caller benchmark¶
Inside daisy’s task library are some common tools used for variant calling. The following guide shows how to install daisy and run a variant-caller benchmark on publicly available data. It starts from scratch, though you will have to have installed a conda environment.
Installation¶
We start by creating a conda environment and installing daisy:
conda create -y -n daisy python=3.6 matplotlib
pip install cgatcore
pip install cgat-daisy
conda install ruamel_yaml pysam
Note
We use a pip install until conda packages is available and are up-to-date.
Next, we install a few variant callers (freebayes, octopus and bcftools) and variant metrics that we want to test:
conda install -y -c bioconda freebayes octopus bcftools samtools bedtools
All of these have already been wrapped in daisy’s Task Library and are ready to be used. Next, we download the aligned exome sequencing data of the NA12878. We will only use chromosome 20 to avoid downloading the whole file (26Gb):
samtools view -h ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/technical/working/20120117_ceu_trio_b37_decoy/CEUTrio.HiSeq.WEx.b37_decoy.NA12878.clean.dedup.recal.20120117.bam 20 | samtools view -bS > NA12878.bam
samtools index NA12878.bam
For variant calling, we will also need the reference genome sequence:
wget ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/phase2_reference_assembly_sequence/hs37d5.fa.gz
gunzip hs37d5.fa
Running the benchmarks¶
Create a benchmark.yml file:
title : >-
Benchmarking variant callers on NA12878 exome data
description: >-
This benchmark calls short variants on NA12878 exome data
evaluates the results by comparison against a truth data set
(Genomes in a Bottle, NA12878).
tags:
- SNV calling
- NA12878
database:
# we will uload results to a local sqlite database
url: sqlite:///./csvdb
setup:
suffix: vcf.gz
tools:
- bcftools
- freebayes
- octopus
metrics:
- bcftools_stats
input:
reference_fasta: hs37d5.fa
bam:
- NA12878.bam
bcftools:
options: --format-fields GQ,GP --multiallelic-caller
bcftools_stats:
options: --fasta-ref hs37d5.fa --apply-filters "PASS,."
# apply a hard filter to freebayes output
task_specific:
freebayes.*:
filter_exclude: "FORMAT/GT == '.' || INFO/DP < 5 || QUAL < 20"
Now run the benchmark:
daisy run -v 5 make all
You can now upload the results to the database:
daisy run -v 5 make upload
This will organize the metric data into an sqlite database. To get the number of variants called, you can query the database:
sqlite3 -header -csv csvdb "select i.tool_name, m.key, m.value
FROM bcftools_stats_summary_numbers AS m, instance AS i
WHERE i.id = m.instance_id AND key='number_of_SNPs' "
which will produce the following output:
| tool_name | key | value |
|---|---|---|
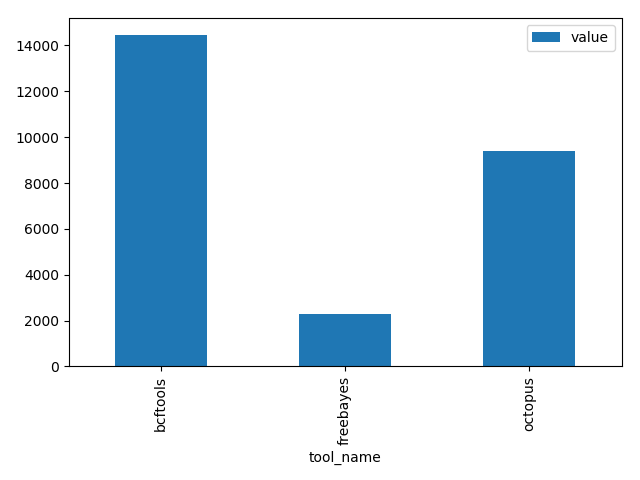
| bcftools | number_of_SNPs | 14454 |
| freebayes | number_of_SNPs | 2308 |
| octopus | number_of_SNPs | 9386 |
Note that such tables can be easily obtained within pandas and used for plotting. For example, the following small python snippet:
import pandas
import sqlalchemy
import matplotlib.pyplot as plt
database = sqlalchemy.create_engine("sqlite:///./csvdb")
df = pandas.read_sql(
"SELECT i.tool_name, m.key, m.value "
"FROM bcftools_stats_summary_numbers AS m, instance AS i "
"WHERE i.id = m.instance_id AND key='number_of_SNPs' ", database).set_index("tool_name")
df.plot.bar()
plt.tight_layout()
plt.savefig("number_variants.png")
will create the following figure:
Adding another metric¶
For a proper variant caller comparison, we should compare against a gold standard of variant calls for our data set. This is available from the NIST/Genome in a bottle initiative:
wget ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/latest/GRCh37/HG001_GRCh37_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz
wget ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/latest/GRCh37/HG001_GRCh37_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz.tbi
wget ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/release/NA12878_HG001/latest/GRCh37/HG001_GRCh37_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_nosomaticdel.bed
Because this is an exome data set, we restrict the high-confidence regions to captured regions:
bedtools genomecov -ibam NA12878.bam -bg | awk '$4 >= 10 ' | bedtools merge -d 10 -i stdin | bgzip > high_coverage_regions.bed.gz
bedtools intersect -a HG001_GRCh37_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_nosomaticdel.bed -b high_coverage_regions.bed.gz | bedtools sort | bgzip > callable_regions.bed.gz
tabix -p bed callable_regions.bed.gz
For the comparison, we will use the vcfeval tool from RealTimeGenomics:
conda install -c bioconda rtg-tools
The toolkit requires its specially formatted reference sequence:
rtg RTG_MEM=16G format -o hs37d5.sdf hs37d5.fa
Now we can amend our benchmark.yml file by adding the rtg_vcfeval
metric to the metrics section:
metrics:
- bcftools_stats
- rtg_vcfeval
The RTG vcfeval tool requires a bit of configuration, so we add the following to benchmark.yml:
rtg_vcfeval:
path: rtg RTG_MEM=16G
map_unknown_genotypes_to_reference: 1
reference_sdf: hs37d5.sdf
reference_vcf: HG001_GRCh37_GIAB_highconf_CG-IllFB-IllGATKHC-Ion-10X-SOLID_CHROM1-X_v.3.3.2_highconf_PGandRTGphasetransfer.vcf.gz
callable_bed: callable_regions.bed.gz
options: --sample=HG001,NA12878 --ref-overlap
We re-run our benchmark:
daisy run -v 5 make all
Note that the variant callers are not re-run, but only additional metrics are computed. Behind the scenes, daisy builds a ruffus workflow which means only tasks that are not up-to-date will be executed. After uploading:
daisy run -v 5 make all
We now have false positive rates and false negative rates in our table:
s3 csvdb "select i.tool_name, m.* from rtg_vcfeval AS m, instance AS i where i.id = m.instance_id "
| tool_name | threshold | true_positive_baseline | true_positive_count | false_positive_count | false_negative_count | false_discovery_rate | false_negative_rate | f_measure | instance_id |
|---|---|---|---|---|---|---|---|---|---|
| bcftools | 12.000 | 1542 | 1542 | 63 | 103 | 0.0393 | 0.0626 | 0.9489 | 4 |
| bcftools | None | 1544 | 1544 | 67 | 101 | 0.0416 | 0.0614 | 0.9484 | 4 |
| freebayes | None | 1607 | 1586 | 167 | 38 | 0.0953 | 0.0231 | 0.9394 | 5 |
| octopus | 5.000 | 1518 | 1518 | 25 | 127 | 0.0162 | 0.0772 | 0.9523 | 6 |
| octopus | None | 1518 | 1518 | 26 | 127 | 0.0168 | 0.0772 | 0.952 | 6 |
Next steps¶
The following are some advanced features not covered in this tutorial:
- Running on multiple data sets within the same benchmark.
- Aggregation of data sets, for example for trio analysis.
- Running time-series analysis for monitoring tool performance.